







Mitochondrial retinopathies are a class of inherited retinal disorders (IRDs) that cause vision loss through retinal dystrophy or optic atrophy, often with syndromic presentations, because of deleterious variants present in the DNA of mitochondria.1 The etiology of several mitochondrial retinopathies has been linked to oxidative stress from impaired oxidative phosphorylation at the level of the retinal pigment epithelium (RPE).1,2
One of the most common mitochondrial DNA sequence variants that can result in IRD involves the MT-TL1 (mitochondrially encoded tRNA leucine 1; OMIM 590050) gene, which encodes for a transfer RNA (tRNA) designated as tRNALeu(UUR) responsible for the addition of amino acid leucine (Leu) during protein synthesis.1,2
The m.3243A>G variant of the MT-TL1 gene has been linked to cytochrome c oxidase deficiency, a condition characterized by the presence of ragged red fibers and skeletal muscle atrophy.3 This variant notably has been associated with various mitochondrial syndromes. Depending on the degree of heteroplasmy, it may lead to conditions such as mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS); Kearns-Sayre Syndrome (KSS); maternally inherited diabetes and deafness (MIDD); as well as other disorders.1-4 Ocular manifestations of the m.3243A>G variant may include ophthalmoplegia, particularly in KSS, and pattern macular dystrophy with varying degrees of vision loss as in MIDD.2-4
Before the 2000s, detection of mitochondrial IRDs required a combination of clinical, biochemical, and imaging findings; however, these methods were not sensitive enough to confirm the specific diagnosis.5 Genetic testing for mitochondrial retinopathies are increasingly becoming available through commercial gene testing panels, including the My Retina Tracker Registry panel (Blueprint Genetics; sponsored by the Foundation Fighting Blindness) and the Inherited Retinal Disorders panel (Invitae Corporation).6 Genetic testing may enable accurate diagnosis, as well as identification of at-risk individuals to facilitate participation in clinical trials and allow, if possible, early intervention and prevention of vision loss.6 Furthermore, it allows diagnosis and management of systemically associated conditions. Additionally, de-identified patient data may be collected in IRD gene registries, such as the My Retina Tracker registry, a research database of people and families affected by rare IRDs, which may serve as data sources to learn about the prevalence of various IRDs, their natural histories, and the impacts of various interventions.6
CASE PRESENTATION
A 57-year-old female patient initially established care for routine eye examination. She had a history of difficulty seeing in low-light conditions. The patient’s medical history was notable for well-controlled late-onset diabetes mellitus (DM) beginning at age 21 that was treated with insulin, along with gastroesophageal reflux disease (GERD) and gastroparesis. She also had bilateral sensorineural hearing loss 7 months after a motor vehicle collision that had caused facial fractures. The patient noted a family history of coronary artery disease in her father and DM in her father, brother, and son; the son was also noted to have late onset DM at age 27. The patient denied family history of retinal disease. She also has a daughter who does not have any significant medical history.
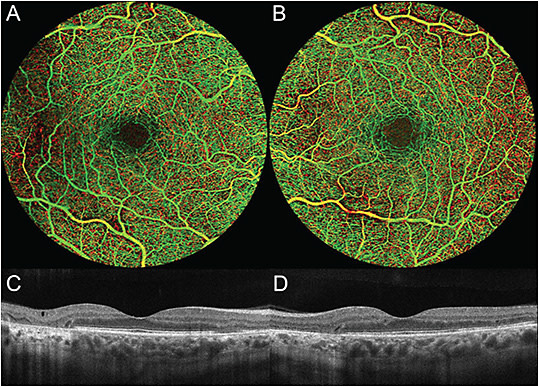
On examination, the patient was noted to have bilateral hyperopia and a best-corrected visual acuity (BCVA) of 20/25 in both eyes. Anterior segment exam was notable for nuclear sclerosis in both eyes. Posterior segment exam showed RPE changes with areas of RPE atrophy in the macula and around the optic disc in both eyes (Figure 1, A and B). Fundus autofluorescence showed a granular pattern of hyper- and hypoautofluorescence surrounding the macula and optic nerve with a characteristic radiating spoke pattern of hyperautofluorescence, as well as patches of hypoautofluorescence characteristic of RPE atrophy (Figure 1, C and D). OCT confirmed outer retinal atrophy (Figure 2, C and D), while OCT angiography showed no evidence of diabetic retinopathy (DR) (Figure 2, A and B).

Multifocal electroretinogram (mfERG) of both eyes revealed abnormal macular function with central depression and irregular response densities. The full-field electroretinogram (ffERG) had only a slight delay in the b-wave of the photopic ERG but had otherwise normal scotopic and photopic pattern of responses. Dark adaptation testing revealed normal kinetics for both cone and rod adaptation, with a rod intercept time of 5.28 minutes in the right eye and 5.66 minutes in the left eye. On a later urologic exam, the patient was found to have gross hematuria.
A retinal dystrophy panel was performed through the My Retina Tracker Genetic Testing Program, which revealed a specific sequence variant causing a pathogenic base substitution in the MT-TL1 gene (m.3243A>G; rs199474657) with a level of heteroplasmy of 18%. The patient’s RPE changes and macular dystrophy, as well as her history of DM and sensorineural hearing loss, were deemed consistent with a diagnosis of MIDD, and her gastroparesis and hematuria could potentially be explained by the MT-TL1 variant as well. The patient was counselled for genetic testing for her children and her family and recommended to undergo further evaluation with cardiology and neurology.
DISCUSSION
The m.3243A>G variant is associated with several mitochondrial syndromes, including, but not limited to, MELAS, KSS, MIDD, and others.1-4 MELAS is characterized by stroke-like episodes before age 40, seizures, dementia, lactic acidosis with low exercise tolerance, and ragged red fibers (RRF) on muscle biopsy.3,7 KSS is characterized by progressive external ophthalmoplegia, pigmentary retinal changes, poor cardiac conduction, and increased cerebrospinal fluid (CSF) protein.2,4,8 MIDD is characterized by adult-onset DM and sensorineural hearing loss, with common ocular manifestations including pigmentary retinal changes and pattern macular dystrophy.2,8 Other potential ocular manifestations include myopia, cataracts, nyctalopia, and central vision loss, and other potential systemic manifestations include constipation and glomerulonephritis.2,8-10 Interestingly, they are reported to have less severe DR compared to the general DM population. Additionally, it is common to find depression on mfERG and diminished b-wave sensitivity on ffERG; loss of b-wave sensitivity is more common than loss of b-wave amplitude in mitochondrial disease, whereas the opposite is true for other retinal degenerative disorders.9,11
In philosophy, Occam’s razor refers to the problem-solving principle that recommends finding explanations constructed with the smallest possible set of elements, also known as the law of parsimony. Our patient’s history of adult-onset DM requiring insulin, along with observed patchy RPE changes and atrophy, aligns with a diagnosis of MIDD. Notably, her macular findings corresponded to grade 2-3 maculopathy associated with the m.3243A>G variant, a hallmark of MIDD.12 The patient also presented with bilateral sensorineural hearing loss, a common feature in MIDD. Although the hearing loss could be attributed to facial trauma sustained during the motor vehicle collision, the concurrent presence of other syndromic features of MIDD suggests that the MT-TL1 variant is the likely cause. Furthermore, the patient’s history of gastroparesis and hematuria can be plausibly linked to mitochondrial dysfunction.
Additionally, the mfERG depression and delays in the b-wave of the photopic ffERG are consistent with the literature findings in MIDD. However, our patient was noted to be hyperopic as opposed to myopic. The patient’s history does not show evidence of other syndromes, such as MELAS or KSS.
The patient denied any retinal diseases in her family history. Her son was noted to have late-onset DM, but has no other symptoms; however, he is at risk of developing other syndromic symptoms, such as sensorineural hearing loss and maculopathy. The patient’s daughter does not have any symptoms related to mitochondrial disease but is similarly at increased risk. The patient’s father and brother have a history of DM as well, but considering that mitochondrial DNA is maternally inherited, they are no more likely than the general population to have the m.3243A>G variant. Additionally, the patient’s mother does not have a history of DM, and it is unclear whether she had symptoms correlating with any of the other syndromes. It is thus possible that the patient’s condition is maternally inherited, but her phenotype more severe due to her level of heteroplasmy, which may vary by individual, or represent a de novo mutation.12
Genetic testing is a significant step in the diagnosis and treatment of mitochondrial retinopathies. Prior to availability of genetic testing, diagnosis would require a combination of clinical, histochemical, pathological, and imaging findings; patients who could not be classified into one of the known syndromes were classified into Complex I, II, III, IV, or V disease.5 Genetic testing became available in the mid-1990s via Sanger sequencing, which enabled discovery of novel variants in patients with mitochondrial disease, but had limited sensitivity for variants in mitochondrial DNA (mtDNA) in a clinical setting.5 Next-generation sequencing (NGS) allowed for increased detection of mitochondrial disease, and by 2010, NGS panels became commercially available; the advent of whole-exome sequencing (WES) augmented the diagnostic abilities of NGS panels.5 Genetic testing for IRDs, including mitochondrial IRDs, are now available commercially through panels such as My Retina Tracker and ID your IRD panels.6 The My Retina Tracker Program panel, provided by Blueprint Genetics, is a carefully curated 351-gene panel targeting relevant genes associated with IRD and includes the entire mitochondrial genome.13
Genetic testing can allow for early identification of variants in at-risk populations, including prior to the development of any symptoms; allow physicians to prevent or delay onset of symptoms and treat treatable manifestations; and enable patients to plan for their future in terms of career and planning a family.6,14 One emerging treatment to prevent transmission of mitochondrial retinopathies is mitochondrial donation therapy (MDT), wherein a donor with healthy mitochondria can donate their mitochondria while preserving the nuclear DNA of the parents.14 MDT has been legal in the United Kingdom since 2015; in the United States, the US Food and Drug Administration (FDA) has so far been prohibited from accepting applications for clinical trials for MDT research.15,16
Nevertheless, while MDT techniques currently provide women affected by mitochondrial retinopathies the chance to have children without transmitting the variant, future advancements in technology may offer an alternative through direct gene editing at the mitochondrial level.
Finally, it is possible for data to be collected in registries (with patient consent and de-identification) to enable further research in disease epidemiology, natural history, and therapy efficacy.6,17 Registries for mitochondrial disease that already exist in the United States include the My Retina Tracker Registry, the North American Mitochondrial Disease Consortium Registry, and the Champ Foundation Registry; there are several additional registries internationally.6,16 NRP
REFERENCES
- Zeviani M, Carelli V. Mitochondrial retinopathies. Int J Mol Sci. 2021;23(1):210. doi:10.3390/ijms23010210
- Oh JK, Lima de Carvalho JR Jr, Nuzbrokh Y, et al. Retinal manifestations of mitochondrial oxidative phosphorylation disorders. Invest Ophthalmol Vis Sci. 2020;61(12):12. doi:10.1167/iovs.61.12.12
- Moraes CT, Ricci E, Bonilla E, DiMauro S, Schon EA. The mitochondrial tRNALeu(UUR) mutation in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS): genetic, biochemical, and morphological correlations in skeletal muscle. Am J Hum Genet. 1992;50(5):934-949.
- Mosewich RK, Donat JR, DiMauro S, et al. The syndrome of mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes presenting without stroke. Arch Neurol. 1993;50(3):275-278. doi:10.1001/archneur.1993.00540030041012
- Saneto RP. Mitochondrial diseases: expanding the diagnosis in the era of genetic testing. J Transl Genet Genom. 2020;4:384-428. doi:10.20517/jtgg.2020.40
- Mathias M. The ins and outs of genetic testing for inherited retinal diseases. Retina Today. 2021;16(5):26-28. https://retinatoday.com/articles/2021-july-aug/the-ins-and-outs-of-genetic-testing-for-inherited-retinal-diseases
- Hirano M. Mitochondrial diseases. In: Brust JM, ed. Current Diagnosis and Treatment: Neurology. 3rd ed. McGraw Hill; 2019:399-406.
- Smith PR, Bain SC, Good PA, et al. Pigmentary retinal dystrophy and the syndrome of maternally inherited diabetes and deafness caused by the mitochondrial DNA 3243 tRNALeu A to G mutation. Ophthalmology. 1999;106(6):1101-1108. doi:10.1016/S0161-6420(99)90244-0
- Latkany P, Ciulla TA, Cacchillo PF, Malkoff MD. Mitochondrial maculopathy: geographic atrophy of the macula in the MELAS associated A to G 3243 mitochondrial DNA point mutation. Am J Ophthalmol. 1999;128(1):112-114. doi:10.1016/s0002-9394(99)00057-4
- Maternally inherited diabetes and deafness. Genetics and Rare Diseases Information Center, National Institutes of Health. Updated February 2023. Accessed September 21, 2023. https://rarediseases.info.nih.gov/diseases/4003/maternally-inherited-diabetes-and-deafness
- Lock JH, Irani NK, Newman NJ. Neuro-ophthalmic manifestations of mitochondrial disorders and their management. Taiwan J Ophthalmol. 2020;11(1):39-52. doi:10.4103/tjo.tjo_68_20
- de Laat P, Smeitink JAM, Janssen MCH, Keunen JEE, Boon CJF. Mitochondrial retinal dystrophy associated with the m.3243A>G mutation. Ophthalmology. 2013;120(12):2684-2696. doi:10.1016/j.ophtha.2013.05.013
- My Retina Tracker Genetic Testing Program. Blueprint Genetics. Updated March 2023. Accessed Sept 21, 2023. https://blueprintgenetics.com/my-retina-tracker-program/
- Stone EM, Aldave AJ, Drack AV, et al. Recommendations for genetic testing of inherited eye diseases: report of the American Academy of Ophthalmology Task Force on genetic testing. Ophthalmology. 2012;119(11):2408-2410. doi:10.1016/j.ophtha.2012.05.047
- Tinker RJ, Lim AZ, Stefanetti RJ, McFarland R. Current and emerging clinical treatment in mitochondrial disease. Mol Diagn Ther. 2021;25(2):181-206. doi:10.1007/s40291-020-00510-6
- Advisory on legal restrictions on the use of mitochondrial replacement techniques to introduce donor mitochondria into reproductive cells intended for transfer into a human recipient. US Food and Drug Administration. Updated March 16, 2018. Accessed September 21, 2023. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/advisory-legal-restrictions-use-mitochondrial-replacement-techniques-introduce-donor-mitochondria
- Abdul-Fatah A, Esmaeilisaraji L, Juan CM, Holcik M. Mitochondrial disease registries worldwide: a scoping review. PLoS One. 2022;17(10):e0276883. doi:10.1371/journal.pone.0276883