







A 10-year-old boy presented as a second opinion from another retina specialist for bilateral iridocyclitis and evaluation to start immunomodulating therapy. He initially presented to his local retina specialist with bilateral decreased vision. Per his initial exam, he was found to have trace flare but no cell on exam, and no other abnormalities on posterior segment exam.
Optical coherence tomography (OCT) revealed bilateral cystoid macular edema (CME). He was initially treated with topical steroids, and later with bilateral dexamethasone intravitreal implants, neither which showed efficacy. His initial uveitis work-up revealed negative results for rheumatoid factor, antinuclear antibody, Quantiferon-TB Gold, human leukocyte antigen (HLA) B27, serum angiotensin converting enzyme, and rapid plasma reagin (RPR).
PRESENTATION + EXAM
On history and review of symptoms, the patient did not endorse any typical signs or symptoms of pediatric uveitis, including photophobia, joint pain or swelling, or rash. Visual acuity was 20/50 both eyes. Intraocular pressure was elevated at 37 mm Hg in the right eye and 34 mm Hg in the left eye. Slit-lamp biomicroscopy was normal, including no cell in the anterior chamber.
Posteriorly, both eyes symmetrically exhibited central macular edema with loss of foveal reflex, and few, scattered bony spicules with uniform white circular flecks in the inferior periphery (Figure 1A).
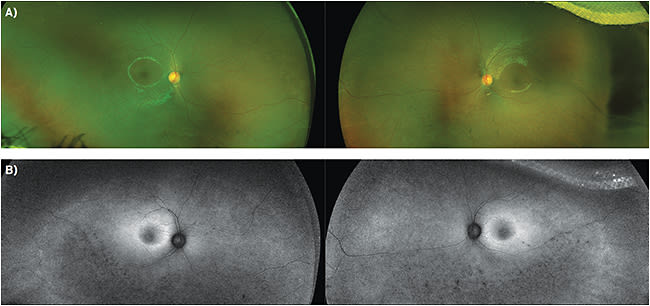
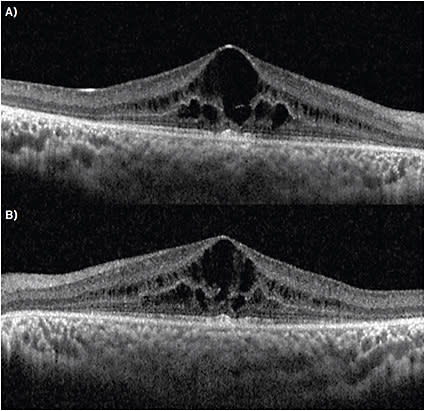
OCT revealed center-involving CME with ellipsoid zone and outer retinal atrophy surrounding the macular edema (Figure 2). Autofluorescence imaging showed blocked fluorescence corresponding to both the bone spicules and hypopigmented circular flecks noted on exam, as well as hyperfluorescence in the peripheral macula.
Based on his exam and imaging, he was thought to have had a variant of retinitis pigmentosa (specifically, an early presentation of retinitis punctate albescens) instead of iridocyclitis. All steroid usage was discontinued at this point and the patient was started on oral acetazolamide and topical dorzolamide for his CME. He is scheduled for an electroretinogram (ERG) in the near future.
DISCUSSION
Retinitis pigmentosa (RP) is broadly defined as an extensive set of diseases of progressive visual dysfunction due to photoreceptor loss.1 These dystrophies are usually hereditary, but the many genetic variants of retinitis pigmentosa mean there are many types of hereditary patterns for RP patients. Thus, the age of presentation for RP patients vary widely, from the first to second decade to later in adulthood. Diffusely affecting both the photoreceptors and the retinal pigment epithelium, these diseases may begin either in the periphery or macula and progress over time.
Most forms of RP result in the death of rod photoreceptors first (leading to poor scotopic and peripheral vision), before eventually affecting cone photoreceptors (progressing to ‘tunnel vision’). With the loss of rod photoreceptors, patients notice nyctalopia and eventual visual field constriction as photoreceptors are progressively degraded. There is often noted to be a poor correlation between the visual acuity and this extent of their tunnel vision.
The earliest typical exam findings include arteriolar narrowing, thought to be due to decreased metabolic demand from the degenerating retina. Later findings include the classic triad of posterior subcapsular cataracts, retinal “bone spicule” pigmentation, and waxy nerve pallor from reactive gliosis.1-4
Cystoid macular edema, which may be a “final common pathway,” is another cause of vision loss in RP.5 The bone spicule pigmentation is theorized to be due to retinal pigment epithelium (RPE) proliferation that occurs as the outer retina thins and attenuated retinal vessels that are now in closer proximity to the RPE begin to lose vascular integrity.6
The RPE undergoes hyperplasia to “plug up” breakdowns in retinal arterial vessels, hence the bone spicule shape. However, the absence of these findings does not rule out retinitis pigmentosa.
Variants of RP may present without any specific exam findings, such as retinitis sine pigmentosa, or with other unusual findings. In this case, the uniform yellow-white hypopigmented circular flecks are concerning for retinitis punctata albescens.
DIAGNOSIS + TRACKING
Diagnosis is made by physical exam in conjunction with office testing. Full field (Ganzfield) ERG is very sensitive to mild photoreceptor dysfunction, and is the gold standard in confirming the diagnosis (especially in suspected cases without typical exam findings). ERG can help confirm the diagnosis and track progression.
In early presentations, decreased a- and b-wave amplitudes are noted. 30 Hz flicker times may be normal or prolonged. As the disease progresses, the amplitudes decrease until the wave is flat, or extinguished.
Goldmann perimetry is used to track peripheral field changes, as patients may respond inadequately to automated perimetry. Color vision is assessed with the Farnsworth D-15 panel. Fundus autofluorescence is helpful, as it has been found to demonstrate pathognomonic patterns found in specific retinal disease states. The autofluorescent lipofuscin patterns have been found to correlate well with OCT and can be used to track progression (Figure 1B). OCT shows structural changes including reduced retinal thickness from photoreceptor loss as well as increased retinal thickness from CME. Genetic testing can support the diagnosis and determine if genetic counseling is necessary.
TREATMENT
Treatment is typically supportive. Posterior subcapsular cataracts can be managed surgically via phacoemulsification, and CME may be responsive to topical or systemic carbonic anhydrase inhibitors, with intravitreal steroids and anti-VEGFs potentially useful for recalcitrant cases. Vitamin supplementation, including vitamin A, docosahexaenoic acid (DHA), lutein, and vitamin E, have not been demonstrated to show clear benefit; in fact, there is evidence excessive vitamin E can be harmful.7-16
UV- and blue-light blocking lenses are advised, though no evidence has shown clear benefit. For advanced disease, low vision examination and therapy may help patients maximize remaining useful vision.
New treatments for RP include genetic therapy and visual prosthetic surgery. Gene therapy using adeno-associated virus (AAV) vector therapy has shown promise for Leber’s Congenital Amaurosis, but given the wide genetic variants of RP, a “one size fits all” treatment using AAV may be difficult.
Visual prosthesis surgery, such as the ARGUS II retinal prosthesis, as well as visual prostheses directly connected to the brain (skipping the visual circuit), are also emerging treatments for end-stage RP.
While bilateral CME is commonly seen in pediatric uveitis, other conditions that can present with bilateral CME must be considered on examination to avoid unnecessary work-up and treatment. In this patient, steroid-induced ocular hypertension was likely caused by his previous steroid treatments; a trial of carbonic anhydrase inhibitors could have been attempted first to avoid this side effect.
Steroid-sparing immunomodulating therapy, a backbone of uveitis treatment, currently has no place in treating RP. We bring this case up to emphasize that while CME is a common finding for uveitis, RP can also present with bilateral CME in pediatric patients, and must be recognized to not only prevent incorrect work-up and treatment, but also to provide proper long-term counseling for such patients. NRP
REFERENCES
- Cukras CA, Zein WM, Caruso RC, Sieving PA. Progressive and ‘stationary’ inherited retinal degenerations. In: Yanoff M, Duker JS, eds. Ophthalmology. 4th ed. Philadelphia, PA: Elsevier; 2014:480-485.
- Levin LA, Albert DM. Ocular Disease: Mechanisms and Management. Saunders/Elsevier; 2010.
- Yanoff M, Sassani JW. Ocular Pathology. 6th ed. Mosby/Elsevier; 2009.
- Yannuzzi, LA. The Retinal Atlas. Saunders/Elsevier; 2010.
- Gorovoy IR, Gallagher DS, Eller AW, Mayercik VA, Friberg TR, Schuman JS. Cystoid macular edema in retinitis pigmentosa patients without associated macular thickening. Semin Ophthalmol. 2013;28(2):79-83.
- Bakthavatchalam M, Lai FHP, Rong SS, Ng DS, Brelen ME. Treatment of cystoid macular edema secondary to retinitis pigmentosa: a systematic review. Surv Ophthalmol. 2018;63(3):329-339.
- Jaissle GB, May CA, van de Pavert SA, et al. Bone spicule pigment formation in retinitis pigmentosa: insights from a mouse model. Graefes Arch Clin Exp Ophthalmol. 2010;248(8):1063-1070.
- Berson EL, Rosner B, Sandberg MA, et al: A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993; 111(6):761-772.
- Massof RW, Finkelstein D: Supplemental vitamin A retards loss of ERG amplitude in retinitis pigmentosa. Arch Ophthalmol. 1993; 111(6):751-754.
- Berson EL, Rosner B, Sandberg MA, et al: Vitamin A supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993; 111(11):1456-1459.
- Berson EL, Rosner B, Sandberg MA, et al: Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol. 2004; 122(9):1297-1305.
- Berson EL, Rosner B, Sandberg MA, et al: Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch Ophthalmol. 2004; 122(9):1306-1314.
- Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128(4):403-411.
- Massof RW, Fishman GA. How strong is the evidence that nutritional supplements slow the progression of retinitis pigmentosa? Arch Ophthalmol. 2010;128(4):493-495.
- Norton EWD. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa [letter to the editor]. Arch Ophthalmol. 1993;111(11):1460.
- Marmor MF. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa [reply letter]. Arch Ophthalmol. 1993;111(11):1460-1461.